This web page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison
What is Phylogeny?
Phylogeny is defined as the history of evolution of a species or group of organisms in the context of a common ancestor (1). Phylogenetic trees are used to show the relatedness between organisms. In order to draw these conclusions, many different algorithms can be performed to to calculate similarities between sequences. BLOSUM Matrix and Percent Identity, two of these techniques, were employed here. In order to compute similarities between sequences you must first align your desired protein sequences. For this study sequences were aligned using ClustalWOmega , see figure 1 below. After alignment, BLOSUM Matrix and Percent Identities were calculated and trees were drawn using the results. (See figures 2-5) Just as there are different algorithms that can be used to calculate similarity, there are different techniques that can be employed to construct tress. The two techniques used in this experiment were Neighbor Joining and Average Distance. A discussion on algorithm and tree construction methodology can be found below with each tree respectively.
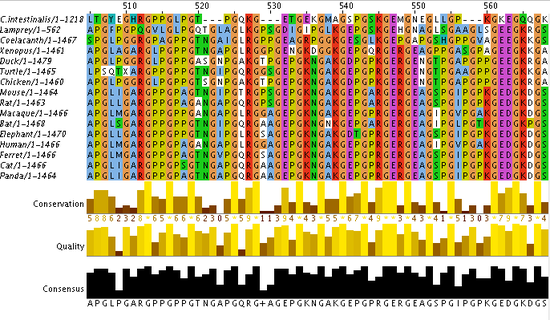
Figure 1: Aligned sequences of organisms used to calculate similarity via BLOSUM Matrix and Percent Identity
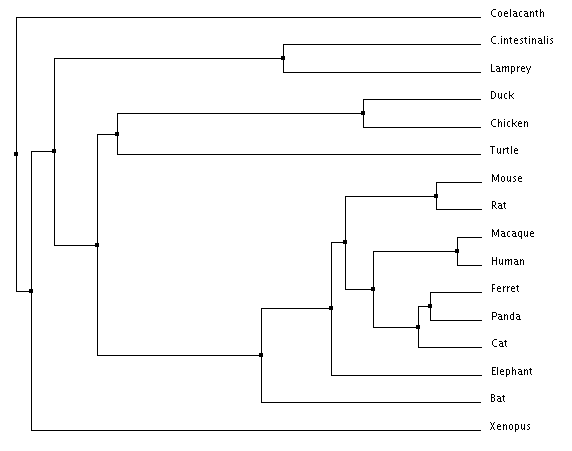
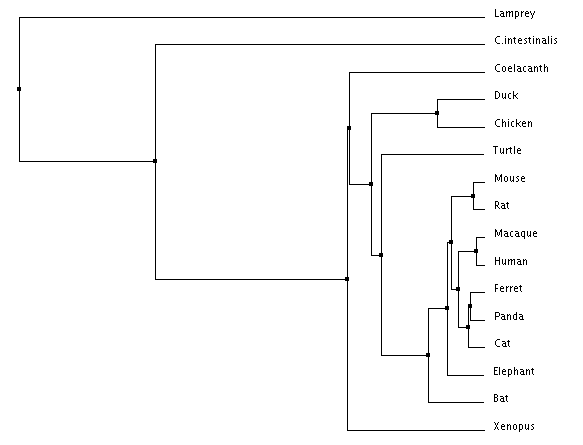
Figure 2: Average distance using Percent Identity Figure 3: Average distance using BLOSUM Matrix
|
|
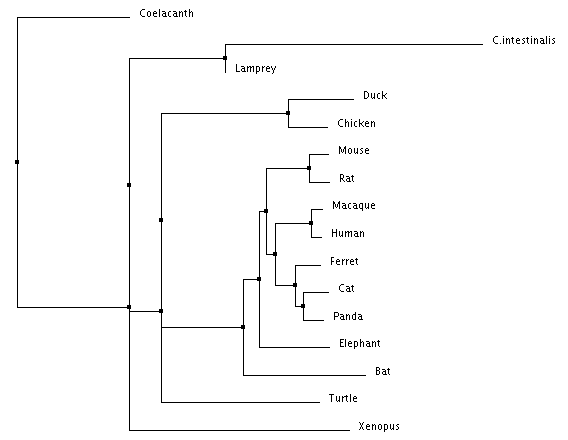
BLOSUM Matrixes and Percent Identity both compare the amino acids between species that were obtained from alignment. BLOSUM matrix calculates a value, called the log-odds score, for each amino acid which indicates the likelihood of two animo acids occurring at the same site by random chance. Percent Identity, on the other hand, simply assigns scores based on if the amino acids are the same or not, neglecting the factors that the log-odds score takes into account. In this experiment both methods were applied to neighbor joining and average distance trees which are discussed in detail below.
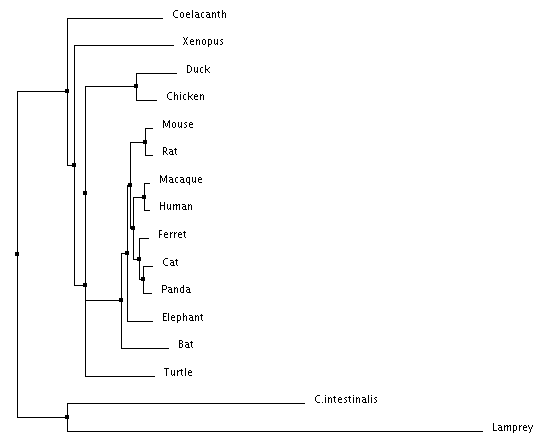
Figure 4: Neighbor Joining using Percent Identity Figure 5: Neighbor Joining using BLOSUM Matrix
|
|
Neighbor joining and and average distance tree construction methods both take into account scores generated from BLOSUM or Percent Identity. Neighbor joining also calculates branch length, while average distance simply connects each species with the same branch length.
Analysis
The most notable difference between the different trees is the different common ancestor obtained from BLOSUM Matrix and Percent Identity calculations respectively. When looking at the average distance trees, Coelacanth is the common ancestor according to Percent Identity while Lamprey is the common ancestor according to the BLOSUM Matrix. This difference highlights the importance of choosing tree construction methods with respect to what you are studying as each method has their own advantages and disadvantages. Animals closely related to humans are in consistent position regardless of algorithm used, and importantly mice, our model organism of interest are consistently related to humans in the same pattern regardless of method.

|

|
Lamprey Coelacanth
References
1) Gittleman, John. "Phylogeny." Encyclopedia Britannica Online. Encyclopedia Britannica, 23 Nov. 2014. Web. 23 Mar. 2015. <http://www.britannica.com/EBchecked/topic/458573/phylogeny>.
2) http://www.ebi.ac.uk/Tools/msa/clustalo/
1) Gittleman, John. "Phylogeny." Encyclopedia Britannica Online. Encyclopedia Britannica, 23 Nov. 2014. Web. 23 Mar. 2015. <http://www.britannica.com/EBchecked/topic/458573/phylogeny>.
2) http://www.ebi.ac.uk/Tools/msa/clustalo/
